User Guide
🔀 Workflow
Tools
TRISTAN is created to discover translating ORFs on the transcriptome using two approaches. One approach evaluates the feasibility of ORFs being translated based on sequence context of the full transcript (TIS Transformer). Another functionality detects translated ORFs based on ribosome profiling data along the transcript (RiboTIE). For a study featuring multiple ribosome samples, it is always recommended to first evaluate ORF scores based on sequence context (TIS Transformer), which will store the predictions for all possible ORFs on the transcriptome within the database. Afterwards, when evaluating ORFs based on ribosome profiling data (RiboTIE), TIS Transformer scores will be included in the result CSV and GTF files.
Tip
Both the TIS Transformer and RiboTIE tools can utilize the same database and config files, where both tools should be run from inside the same project folder.
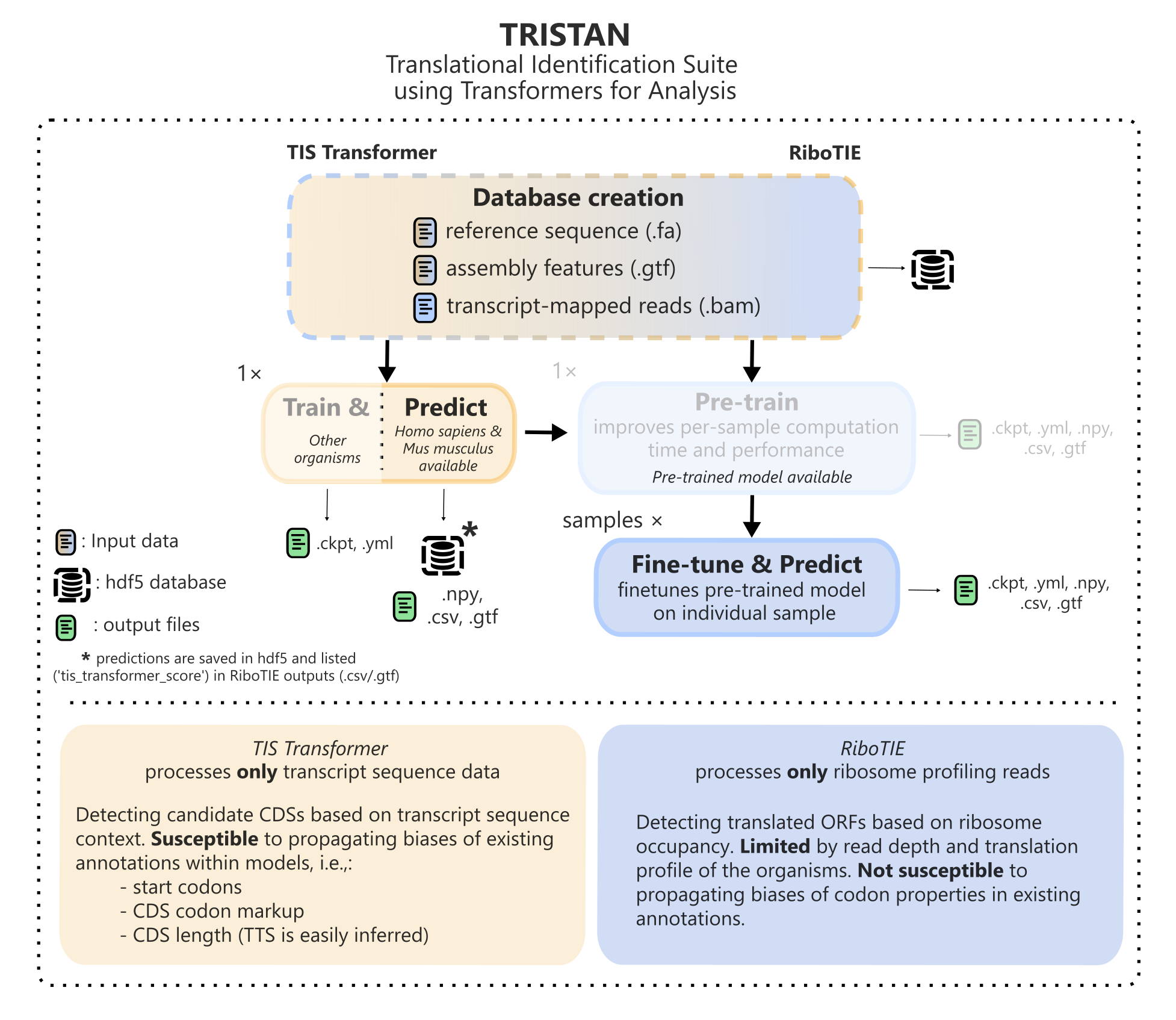
Checkpoint detection
TRISTAN tools automatically resume from certain checkpoints by detecting the presence of output files at certain steps. These can be roughly separated as such:
Parsing data into a HDF5 database
Use --data to only perform data loading. --overwrite_data can be used to overwrite ribosome profiling data. Genome information cannot be overwritten and requires the creation of a new database instead (or deletion of the existing one).
Optimizing a model on the HDF5 database (GPU required)
Once models are optimized, model parameters and information is saved in the output folder (*.cktp, *.yml). --overwrite_models can be used to prevent either TIS Transformer or RiboTIE from utilizing these models and instead fine-tune new models from scratch. Alternatively, see the section on trained models to utilize custom pre-trained models.
Creatinging predictions for a dataset (GPU required)
Multiple models are trained to predict the presence of translated ORFs on non-overlapping folds of the data. Model predictions are saved in numpy (*.npy) files. --overwrite_preds can be used to prevent either TIS Transformer or RiboTIE from utilizing these saved predictions and instead create new predictions. More info on the format of generated numpy arrays can be found here.
Collecting metadata for the top ranking predictions
As a last step, RiboTIE and TIS Transformer collect metadata for the top predictions accross the full transcriptome. Several steps are performed that filter high-scoring ORFs from the final output table, such as those defined by the user, and CDS variant filters.
Tip
Make sure to check out topics on Parallelization and Workflow creation when handling a large number of samples within your project.
⚙️ Config Files
The easiest control of parameters for a given project is through config YAML files. All parameters can be passed using these YAML files, supporting a more intuitive structure for more complex arguments such as lists or dictionaries. Multiple config files can be passed at once in addition to function flags. In case of repeated parameter values, latter values overwrite previous ones, with function flags overwriting config files.
Example config
Tip
A single config file is recommended to control both TIS Transformer and RiboTIE. Parameters not used by either tool are ignored.
The minimal configuration requires paths to the annotation and sequence files, the ribosome profiling data, and a designated path for the output database.
gtf_path: Path to your GTF annotation file.fa_path: Path to your FASTA sequence file.h5_path: Path where the output HDF5 database file will be stored.out_prefix: Specifies a prefix for output prediction files. If not set, it defaults to theh5_path.ribo_paths: A dictionary specifying unique IDs and their corresponding paths to the mapped ribosome profiling data (e.g., in SAM format). These IDs should not be changed after the initial data processing.samples: Allows you to select a custom subset of ribosome profiling data for training or merge technical replicates. You can list the sample IDs directly or group them under a new name. Often used in combination with--parallelto process samples in parallel
gtf_path: gtf_file.gtf
fa_path: fa_file.fa
h5_path: database.h5
out_prefix: "results/db_"
# Add entries for each ribosome profiling dataset
ribo_paths:
SRR000001: path/to/mapped/riboseq.sam
SRR000002: path/to/mapped/riboseq.sam
SRR000003: path/to/mapped/riboseq.sam
# Optional: Select specific samples or merge replicates for training
# Example 1: Use only SRR000001 and SRR000003
samples:
- SRR000001
- SRR000003
# Example 2: Merge SRR000001 and SRR000002 as 'group1'
# samples:
# group1:
# - SRR000001
# - SRR000002
# group2:
# - SRR000003
Way more advanced config
Check out tis_transformer --help or ribotie --help for all supported function flags.
Trained models
After training a set of models, TRISTAN will create *.ckpt files and an accompanying model configuration file (*_params.<tool_prefix>.yml). These configuration files contain all parameters necessary to apply this model for prediction purposes. To do so, list the parameter config file alongside the default config file:
ribotie default.yml <*_params.yml>
This informs the tool how to apply fine-tuned models, making the tool skip directly to the prediction step. It is necessary for the config file to live within the same folder as the saved *.ckpt files. Importantly, these files contain the allocation of chromosomes for each model, where models are not meant to be be applied on transcripts they were trained on. An example parameter YAML file for models fine-tuned on a ribosome profiling BAM file (RiboTIE) could look like:
lr: 0.0008
patience: 1
trained_model:
folds:
0:
test: []
train: ['3', '5', '7', '11', '13', '15', '19', '21', 'X', 'chr3', 'chr5', 'chr7', 'chr11', 'chr13', 'chr15', 'chr19', 'chr21', 'chrX']
transfer_checkpoint: db_BT245H3K27M_C1_f0.rt.ckpt
val: ['1', '9', '17', 'chr1', 'chr9', 'chr17']
1:
test: ['1', '3', '5', '7', '9', '11', '13', '15', '17', '19', '21', 'X', 'chr1', 'chr3', 'chr5', 'chr7', 'chr9', 'chr11', 'chr13', 'chr15', 'chr17', 'chr19', 'chr21', 'chrX']
train: ['2', '6', '8', '10', '14', '16', '18', '22', 'Y', 'chr2', 'chr6', 'chr8', 'chr10', 'chr14', 'chr16', 'chr18', 'chr22', 'chrY']
transfer_checkpoint: db_BT245H3K27M_C1_f1.rt.ckpt
val: ['4', '12', '20', 'chr4', 'chr12', 'chr20']
test: [] indicates acceptance of all chromosomes to be valid for prediction excluding those listed in ‘train’ and ‘val’. This approach allows evaluation of chromosome names not observed during training of the models, improving compatibility of trained models with databases that include plasmids or other custom FASTA sequences.
Attention
When applying references that adhere to different naming, make sure to update the yml files to prevent that all transcripts are processed by the db_BT245H3K27M_C1_f0.rt.ckpt model as none of the chromosome names overlap and are thus assigned to the wildcard group []. Check the output log to evaluate chromosome assignment.
📝 Results
Output Files
In all cases, all potential translation initiation sites on the transcriptome are returned and stored in *.npy files. However, providing detailed information for millions of predictions isn’t practical or useful.
Therefore, we focus on generating user-friendly output for predictions that meet specific criteria. For these selected predictions, the tools will generate the following output files:
Raw Predictions:
<out_prefix>_<sample>.npyStandard Results:
CSV format:
<out_prefix>_<sample>.csvGTF format:
<out_prefix>_<sample>.gtf
Novel Coding Sequences:
CSV format:
<out_prefix>_<sample>.novel.csvGTF format:
<out_prefix>_<sample>.novel.gtf
Result Table Without CDS Variant Filtering:
CSV format:
<out_prefix>_<sample>.redundant.csvGTF format:
<out_prefix>_<sample>.redundant.gtf
Optimized Model (if applicable):
ckpt file:
<out_prefix>_<fold>.<tool_prefix>.ckptmodel params:
<out_prefix>_params.<tool_prefix>.yml
GTF files can be applied with tools such as gffread to extract sequences etcetera. You can customize the conditions for selecting called ORFs when generating the results tables. If you need to regenerate these tables, simply use the --overwrite_results flag.
For example, to include all possible start codons in your results, you would run:
ribotie <config.yml> --start_codons ".*" --overwrite_results
ORF property filters
Custom filtered can be toggled or altered by the user. Top scoring ORFs are filtered on:
--prob_cutoff: the model prediction cutoff score to separate the positive and negative set. Default settings are 0.125 for RiboTIE and 0.03 for TIS Transformer. Differences betwee thresholds are explained by differences in precision and expected positive calls for each tool.--start_codons: allowed start codons (start_codon). Default settings filter out non-near-cognate (*TG) codons. This flag expects a regular expression with the allowed start codons. Default value is".*TG$"--include_invalid_TTS: boolean flag indicating a valid translation termination site (TTS_on_transcript) is not required. Default setting isFalse.
CDS variant filters
Reads mapped to the genome are assigned to all transcripts that share the 5’ genomic site of the read. For genes featuring multiple transcript isoforms, ribosome fingerprint of a coding sequence can thus be (largely) shared, which results in ORF calls across multiple transcripts that are likely false positives. To illustrate, without post-processing, a uORF on one transcript could have an identical genomic region as an existing annotated CDS on another transcript (see CDS clone). To address multiplicative calls rising from this property of ribosome profiling, we now provide a filtered down list of ORF calls following these listed strategies
For ORF calls (positive set; adjustable by threshold) with identical genomic TIS coordinates, a single ORF is selected using the following priority order (if not first excluded by other filters):
Annotated CDS – (custom filters)
Truncation/Extension – (custom filters & CDS clones)
d/u(o)ORF/intORF/lncRNA-ORF – (custom & CDS-region filters)
varRNA-ORF – (custom & CDS-region filters)
Where ‘protein coding’ takes precedence over other transcript biotypes in case multiple transcript biotypes exist within the same group. Here, varRNA-ORFs denote ORFs on transcript other than ‘protein coding’ and ‘lncRNA’. Called ORFs can be variant CDSs present on alternate transcript biotypes, such as ‘retained intron’, ‘nonsense mediated decay’, or ‘processed pseudogene’.
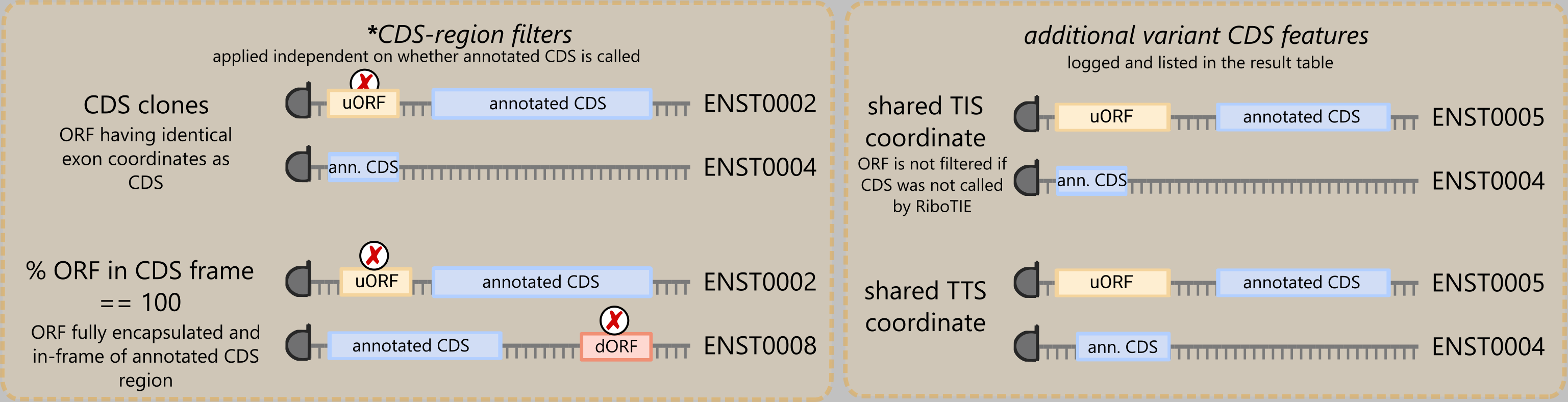
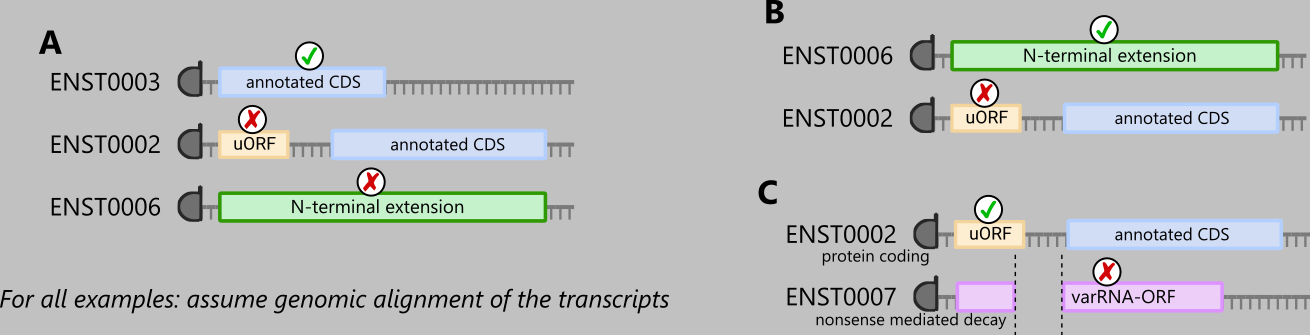
Some example scenarios include, remember that filtering only occurs across groups of called ORFs sharing the same TIS:
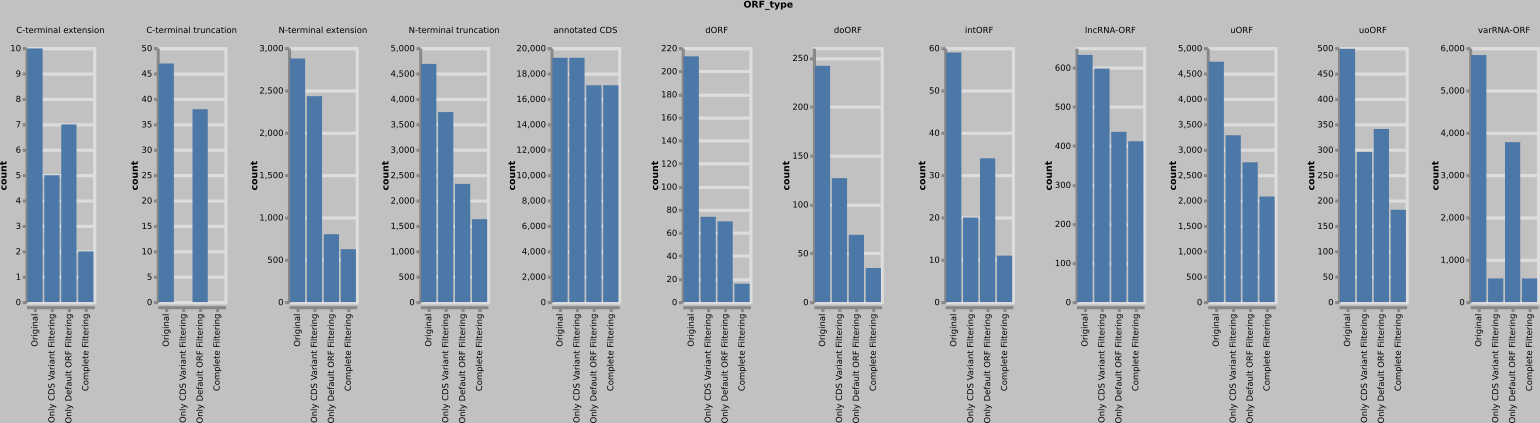
A large number of called truncations/extensions and ncORFs are rejected following the above filter methods due to having shared genomic coordinates features to annotated CDSs. Overall, lncRNA-ORFs are the least affected. The same trends are observed on all evaluated data. An example dataset shows the following numbers:
Near-miss identifier
RiboTIE, unlike previous tools processing ribosome profiling data, does not create ORF libraries or has access to start codon information when making predictions. Essentially, it only parses ribosome profiling information along the transcript.
It is observed that, for transcripts featuring fewer mapped reads around the translation initiation site, RiboTIE is more prone to miss translation initiation sites by several bases. To address this issue, a neighborhood searching step is performed when creating the result table that corrects non-ATG predictions to in-frame ATG positions if present within a 9 codons distance. Performed corrections are listed as correction in the result table. The maximum distance between the non-ATG site and nearest ATG site can be altered using the --distance flag. Alternatively, this feature can be disabled using the --no-correction flag.
MultiQC
RiboTIE now generates several report files that are automatically detected by MultiQC and displayed when running it in a parent directory.
🌳 Effeciency Tips
Both TIS Transformer and RiboTIE require GPU usage, which is expensive and typically limited. After running either tool, optimized models are now saved under <output_prefix>_<fold>.<tool_prefix>.ckpt and accompanied by a <output_prefix>_params.<tool_prefix>.yml file. In addition, we provide two pre-trained models for both homo_sapiens and mus_musculus organisms that allow the majority of users to apply TIS Transformer without training a new model.
When training a model, it is recommended to save these files, as training on the genome sequence (i.e., TIS Transformer) or sample expression data (i.e., RiboTIE) should only be done once ever. Even if a newer genome assembly is released, and even when reads are newly aligned to updated reference genomes, it is not necessary to re-run the training process of either tool. To apply the fine-tuned models on the new data, simply recreate a database with updated data and pass the generated <output_prefix>_params.<tool_prefix>.yml config file. e.g. ribotie <default_config.yml> <model_params.yml>
The main issue that can arise when applying optimized models on new data is the alternate naming of the chromosomes (e.g. ENSEMBL vs. REFSEQ), in which case it is necessary to manually add/change these in the YAML file. See the section on trained models for more information.
⚡️ Parallelization
The use of a single hdf5 database has limitations towards upscaling and parallelization of RiboTIE. In many cases, it is desired to process multiple ribo-seq samples in parallel.While samples are processed independently, having multiple processes write and read to and from a single hdf5 can result in I/O errors.
--parallel creates independent hdf5 databases for each ribo-seq sample ({h5_path%.h5}_{ribo_id}.h5), allowing individual samples being processed in parallel when multiple GPUs are available. However, succesful use of --parallel requires execution of commands in the following order:
tis_transformer <config.yaml> --data: parse only genomic features in a single mainhdf5databaseribotie <config.yaml> --parallel --samples <ribo_id>: parse the ribo-seq data of a single sample into a separatehdf5database and process it. This argument can be ran in parallel for multiple gpus. Check out--devicesif running on a single system rather than a HPC infrastructure.
Here, the --samples function flag can be used to overwrite the samples listed in the default configuration file to ensure only a single sample (or group of samples) is processed.
Attention
RiboTIE still pulls data from the hdf5 database containing the genomic features when processing the result table, so make sure these databases always exist under the same folder as defined by the --h5_path argument.
🐍 Snakemake
Snakemake or other workflow software can be a useful tool to navigate the various steps performed using the TRISTAN software. We give some examples here to help navigate setting up your pipeline.
# from config.yml, can also be part of a snakemake configuration file
samples = {
"sample_1": "sam_data/SRR000001.sam",
"sample_2": "sam_data/SRR000001.sam",
"sample_3": "sam_data/SRR000001.sam"
}
rule all:
input:
expand("test_out/{sample}.npy", sample=samples.keys())
# `tis_transformer --data` is called first as running multiple `ribotie` commands in parallel
# would result in parallel processes parsing the genome assembly features, which is only required
# once (and stored under `{h5_path}` and within the backup location).
rule create_tristan_config:
localrule: True
output:
"config.yml"
params:
gtf=config['gtf_path'],
fa=config['fa_path'],
out_prefix=config['out_prefix'],
run:
config_dict = {
"gtf_path" : params.gtf,
"fa_path" : params.fa,
"out_prefix" : params.out_prefix,
"h5_path" : "GRCh38v113.h5",
}
with open(f"config.yml", 'w') as outfile:
yaml.dump(config_dict, outfile, default_flow_style=False, sort_keys=False)
rule tristan_parse_genomic_features:
input:
"config.yml"
output:
"GRCh38v113.h5"
resources:
mem_mb=21000,
slurm_partition="standard",
shell:
"""
tis_transformer {input} --data
"""
# This is recommended to include tis_transformer scores in subsequent RiboTIE results retrieved from
# this database.
rule tis_transformer_predict:
input:
config="config.yml",
# ancient is used as db get's updated after predictions are performed
db=ancient("GRCh38v113.h5"),
output:
csv="GRCh38v113.csv",
gtf="GRCh38v113.gtf"
resources:
mem_mb=21000,
slurm_partition="gpu",
slurm_extra="--gpus=1"
shell:
"""
tis_transformer {input.config} --model human
"""
# Given the high Memory requirements when loading in BAM files, it can be advantageous to create
# an extra rule for parsing the riboseq data separately using a partition that does not require GPU resources
rule ribotie_parse_riboseq_samples:
input:
config="config.yml",
db="GRCh38v113.h5",
mapped=lambda wildcards: samples[wildcards.sample],
# Ensures TIS Transformer is completed first and predictions are in RiboTIE output tables
tis_tr="GRCh38v113.csv"
output:
db="GRCh38v113_{sample}.h5"
# Instead of {params.json}, users can also use --samples {wildcards.sample} if ribo_paths is already defined
# in the config. The outlined approach is more stable in case more samples are added in the future, as recreation
# of the config.yml file to extend ribo_paths would, by default, result in snakemake re-running all TRISTAN rules
# in case the creation of the config.yml file is part of the pipeline.
params:
json = lambda wc: f'{{"{wc.sample}": "{samples[wc.sample]}"}}'
resources:
mem_mb=120000,
slurm_partition="largemem",
shell:
"""
ribotie {input.config} --ribo_paths '{params.json}' --data --parallel
"""
# Run RiboTIE on processed data for individual samples
rule ribotie_finetune_and_predict_riboseq_samples:
input:
config="config.yml",
db="GRCh38v113_{sample}.h5",
mapped=lambda wildcards: samples[wildcards.sample]
output:
csv="GRCh38v113_{sample}.csv",
gtf="GRCh38v113_{sample}.gtf",
model="GRCh38v113_{sample}_params.rt.yml",
params:
json = lambda wc: f'{{"{wc.sample}": "{samples[wc.sample]}"}}'
resources:
mem_mb=21000,
slurm_partition="gpu",
slurm_extra="--gpus=1"
shell:
"""
ribotie {input.config} --ribo_paths '{params.json}' --parallel
"""
When working with StringTIE to determine novel transcripts, one can extend this pipeline to incorporate detection of translated ORFs on these novel transcripts as well. Here, make sure to apply the models trained by the previous steps to save computational resources.
For example, a rule similar to this:
rule tis_transformer_predict_custom_transcripts:
input:
config="config_stringtie.yml",
db=ancient("GRCh38v113_stringtie_extension.h5"),
mapped=lambda wildcards: samples_CST_TS[wildcards.sample],
output:
csv="GRCh38v113_stringtie_extension.csv",
gtf="GRCh38v113_stringtie_extension.gtf",
resources:
mem_mb=21000,
slurm_partition="gpu",
slurm_extra="--gpus=1"
shell:
"""
tis_transformer {input.config} --model human
"""
rule ribotie_predict_custom_transcripts:
input:
config="config_stringtie.yml",
db="GRCh38v113_stringtie_extension_{sample}.h5",
model="GRCh38v113_{sample}_params.rt.yml",
mapped=lambda wildcards: samples_CST_TS[wildcards.sample]
output:
csv="GRCh38v113_stringtie_extension_{sample}.csv",
gtf="GRCh38v113_stringtie_extension_{sample}.gtf",
params:
json = lambda wc: f'{{"{wc.sample}": "{samples_CST_TS[wc.sample]}"}}'
resources:
mem_mb=21000,
slurm_partition="gpu",
slurm_extra="--gpus=1"
shell:
"""
ribotie {input.config} {input.model} --ribo_paths '{params.json}' --parallel
"""